Parte 2:A Teoria da Descendência Comum Universal
"A teoria da evolução de Darwin é basicamente 'descendência com modificação' através da seleção natural na formação de novas espécies. É ponto pacífico que dentro de uma espécie ocorra descendência com modificação.É discutível, porém, que todos os seres descendam gradualmente modificados de uma forma ou de algumas formas originais. Se assim fosse, a história da vida seria representada pelo gráfico da árvore."
---------------------------------------------------------------------------------------------------------------------------------------
A descendência com modificação não foi apenas observada apenas dentro da espécie, mas também em relações interespecíficas.A macroevolução já foi observada diretamente e indiretamente.Origem de uma nova espécie pela evolução já foi vista até em laboratório (Weinberg, et al., 1992) e um tipo de especiação merece atenção porque permite um estudo detalhado. É o caso do fenômeno “espécies-em-anel”, onde vemos diferenças devido a isolamento parcial dando origem a populações que se comportam como espécies separadas.É uma situação na qual duas populações que vivem em um mesma região e não se reproduzem estão conectadas por um anel geográfico de populações que se reproduzem.Este é o caso do pássaro Phylloscopus trochiloides e da salamandra do gênero Ensatina (Pazza,2004).A última possui tantas diferenças, que duas populações desenvolveram estratégias radicalmente diferentes para evitar predadores-uma exibe padrão críptico e outra mimetiza uma espécie de salamandra venenosa.Além disso, existem as evidências da descendência comum baseada em observação indireta. Elas incluem: anatomia comparativa, biologia do desenvolvimento comparativa, estudos comparativos bioquímicos e genéticos; padrões de biogeografia e o registro fóssil.
O que está por trás da afirmação "que todos os seres descendam gradualmente modificados de uma forma ou de algumas formas originais"?O gradualismo não é um mecanismo de mudança evolutiva.E também não é uma afirmação quanto a taxas ou tempo da evolução (Mayr,2005.p.122).Eventos geneticamente graduais são apenas restrições da área de variação biológica esperada entre duas gerações quaisquer quando observamos suas taxas genéticas, taxas morfológicas,etc. Assim, o gradualismo requer apenas restrição a possíveis mecanismos de descendência comum e adaptação, não afirmando nada sobre se a evolução deve ser constante,rápida, ocorrer frequentemente,etc.Acreditar que é "discutível que os seres descendem de um ancestral comum", porque não ser possível a história da vida esteja "representada pelo gráfico da árvore" mostra apenas uma confusão no entendimento de conceitos científicos.É análogo a dizer que "é discutível que a força entre duas massas sempre esteja de acordo com a lei do quadrado do inverso.Se assim fosse, a aceleração de todas as massas estariam de acordo com a existência de um valor exato da constante gravitacional G."A razão de seu comentário está incorreto é que a construção de uma "representação da vida no gráfico da árvore" não é possível, porque valores exatos de mensurações não são sabidos em ciência.Aproximações de mensurações não existem apenas em biologia evolutiva, mas em todas as ciências.O que não é impossível é construir uma árvore filogenética padrão, uma aproximação da verdadeira e única genealogia da vida.O mesmo poderíamos dizer sobre o conhecimento do valor exato da constante gravitacional universal, já que até este tem medidas independentes que desacordam.O que se pode fazer é construir uma constante gravitacional G padrão para que todas as massas sempre se acelerem em um valor específico que nunca seja massivamente inconsistente com ela.De modo similar, o que se pode fazer para testar a descendência comum universal é construir uma árvore filogenética padrão, onde toda filogenia específica seja compatível com ela.
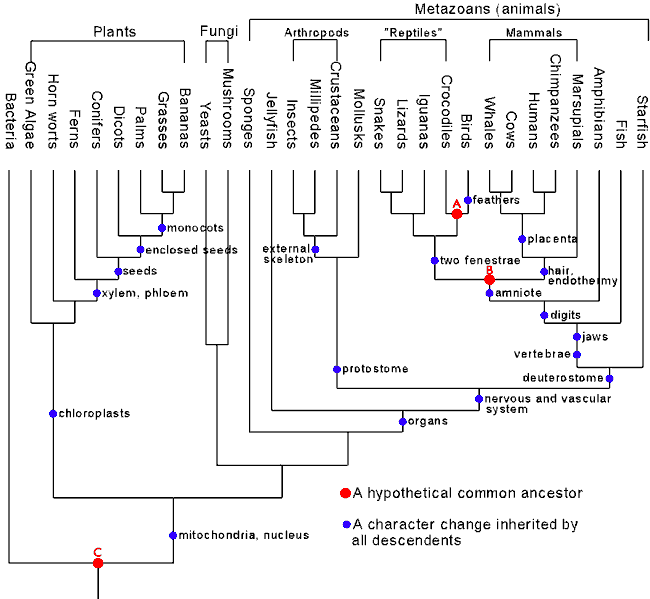
Árvores são construídas a partir de dados, com a utilização de métodos que podem produzir árvores distintas.Se os dados são igualmente consistentes com diversos padrões de ramificação, o julgamento acerca das relações deve ser suspenso.Mas uma filogenia bem determinada pode realizar as predições da teoria da descendência comum universal se todos os métodos devem fornecer o mesmo ramo em um grande (e confiável) conjunto de dados e com cada ramo sendo bem sustentado.Por fim, considerando que qualquer filogenia com tais características é a melhor aproximação de uma verdadeira e única genealogia de todas as espécies, ela forma uma árvore filogenética padrão.Isso significa que apenas uma filogenia assim pode ser uma descrição realista e rigorosa da descendência comum.Um exemplo prático dela é esta árvore neste gráfico criado pelo bioquímico norte-americano Douglas Theobald (2004) no Talk Origins Archive.
Outra questão no teste de filogenias é explicar porque ela não prediz a exata congruência de mensurações filogenéticas construídas por dados independentes (seja por dados moleculares, embriológicos, paleontológicos e/ou morfológicos).Em caso de filogenias envolvendo muitos táxons, o simples fato de colocarmos muitas espécies num cladograma faz com que o número de árvores filogenéticas possíveis cresça fatorialmente, porque a fórmula para se calcular o número de árvores possíveis é NR=(2n-3)!! ou (2n-3)!/(2n-2(n-2)!), onde "n" é o número de táxons no cladograma.No caso de cinco espécies são 105 árvores possíveis.Em caso de 10, são 34.459.425.No caso de 15 espécies, são 213.458.046.676.875 árvores possíveis.Mas se se consegue reduzir o número de árvores prováveis para 34 , isso significa que se está fazendo uma mensuração com uma acurácia de uma parte em 1 milhão (34/34.459.425 árvores possíveis) .Se colocássemos 15 espécies,poderia-se estabelecer centenas de árvores prováveis e a acurácia seria considerada ainda maior.Como não existe mensuração perfeita na obtenção de valores independentes em ciência, cientistas utilizam estatísticas de análise de congruência , para estimar o quanto de discrepância a teoria permite para conflitos de árvores filogenéticas obtidas por dados independentes (sejam eles moleculares, embriológicos, morfológicos, etc.).Um dos cálculos de incongruência filogenética é o valor de P e o método mais popular de avaliação da confiabilidade de árvores é o bootstrapping (ambos serão analisados a seguir).Como se vê na parte 5 deste artigo, a evidência disponível existe um número excepcional de filogenias bem determinadas inferidas por métodos independentes combinando-se com significância estatística.Outra coisa deve ser analisada.Se a descendência comum fosse falsa, existiriam independentes filogenias com forte suporte mostrando nível de incongruência elevada.Como isso ainda não ocorre, percebe-se que ainda não surgiu dados que parecessem um problema sério para a teoria.
Se a descendência com modificação é aceitável dentro da espécie, porque não em relações intraespecíficas?Nunca observamos diretamente que todos os humanos descendendo gradualmente de um ancestral remoto, mas temos evidências impressionantes disso.Idem para a descendência com modificação em relações inter-específicas.
Algumas filogenias são consensuais e tem ramos bem-suportados, de modo que uma inversão nela refutaria a evolução.Por exemplo, a evolução prediz com certeza que humanos já tiveram ancestrais caudados,assim como os primeiros primatas.Taxonomistas englobam humanos, chimpanzés, gorilas, orangotangos e gibões na Superfamília Homonoidea e são justamente esses animais que compartilham a sinapomorfia "ausência de cauda".Podemos citar um exemplo de uma evidência que confirma uma hipótese da filogenia de consenso universal e outro que a contradiga?Sim, a existência de atavismos em bebês humanos que possuem caudas , fato registrado na literatura médica (ver fotos no link).Como isso pode ocorrer? Na cauda embrionária, existente em nós humanos, e em outros macacos, em determinado estágio essas células morrem.Porém, uma mutação faz com que as células da cauda morram um tempo após terem sido formadas. Da mesma forma que uma mutação fixada na maioria da população é responsável pela ausência da cauda na maioria de nós, mutações raras podem fazê-las reaparecer em alguns indivíduos.Os primeiros primatas tinham caudas e a ausência delas é uma característica derivada que é existente em apenas em cinco grupos primatas que representam a Superfamília Hominoidea.Vértebras e cartilagem têm ocasionalmente sido encontradas em caudas humanas. De qualquer forma, vértebras não são um requerimento para caudas. M. sylvanus é um exemplo perfeito de primata cuja cauda carnuda não têm vértebras. Nós agora sambemos os genes responsáveis pelo desenvolvimento das caudas em mamíferos, e que todos os humanos os têm. Muita dessas caudas são complexas estruturas compostas de camadas simétricas de músculos voluntários, vasos sangüíneos, nervos especializados e órgãos sensitivos, são raramente herdadas, ainda que vários casos familiares sejam conhecidos. Em um caso isso foi herdado por pelo menos três gerações de indivíduos do sexo feminino. (Standfast,1992).Não seria indício de ancestralidade comum se fossem homoplasias, mas não são. Os genes existem e produzem a cauda que permanece em todos os animais até um certo período de gestação, quando nos primatas mais modernos elas regridem.Baseado na hipótese da descendência comum e na árvore filogenética padrão , podemos esperar ver caudas atávicas em humanos, mas essa hipótese poderia ser confirmada como falsa se víssemos algum caso de bebê nascer com escamas.
Há evidência até para o caso em que todos estão agrupados num único grande táxon universal.Por exemplo, sabe-se que todos os seres dos cinco reinos tem um genoma baseado em ácidos nucléicos e o mesmo código genético que usam os mesmos códons e aminoácidos porque descendem de um ancestral comum, poderia se encontrar uma evidência contrária falseadora se descobrissem algum organismo com um genoma que não é baseado em ácidos nucléicos ou que utilize o mesmo código genético com códons e aminoácidos diferentes.Uma vez que ter exatamente as mesmas trincas que codificam exatamente os mesmos aminoácidos não é funcionalmente necessário e nem se deve a razões de afinidades químicas , a explicação mais racional é que ele é um "acidente congelado",onde um desvio seria letal.Um organismo que lesse GGC como fenilalanina em vez de glicina, morreria ainda no estágio de ovo (Ridley,2006.p.81).
Por fim, é bom esclarecer que a teoria da descendência comum universal é a única que pode dar conta de explicar cientificamente da unidade, diversidade, e padrões da vida terrestre.Ela pode ser testada independentemente dos mecanismos teóricos.Ela também é considerada fato científico pela maioria dos biólogos, embora algumas teorias evolutivas sobre os mecanismos evolutivos possam não ter o mesmo status.
COMO AS FILOGENIAS SÃO CONSTRUÍDAS E AVALIADAS?
Para entender como a descendência comum é descrita pelas filogenias é necessário compreender a metodologia da sistemática filogenética.A biologia evolutiva está constantemente tentando encontrar grupos monofiléticos.Quando um grupo é considerado monofilético, isso quer dizer que todos os ancestrais daquele grupo são derivados de um ancestral comum, o qual não é ancestral de nenhum outro grupo.A abordagem a sistemática, conhecida atualmente como cladística, foi formulada por Willi Hennig e estabelece que as sinapomorfias - estados de caráter unicamente compartilhados e derivados- são filogeneticamente informativas.Assim, se compararmos o traço “número de dígitos”, ele não seria relevante numa análise filogenética entre humanos, lagartos e cavalos, já que é ancestral a todos os três.Mas se considerarmos cavalos, zebras e humanos ele seria relevante, pois se encontra num estado derivado por cavalos e zebras em relação a humanos.A similaridade primitiva herdada de um ancestral distante é chamada de plesiomorfia.As que são derivadas são chamadas de apomorfias.Uma sinapomorfia em níveis taxonômicos mais altos pode representar uma plesiomorfia desinteressante em níveis inferiores.Um exoesqueleto quintinoso ajuda a distinguir artrópodes de anelídeos e dos moluscos, mas não é útil na distinção entre espécies de borboletas.Se as espécies A, B e C reunem mais traços comuns entre si que D, E e F (e com um postulado ancestral comum mais recente por causa disso), então pelo critério da parcimônia, uma característica em comum compartilhada por D,E e F é provavelmente ancestral.É possível a codificação dos caracteres, de forma que os mesmos possam ser utilizados em um algoritmo, manual e automatizado.Procura-se a árvore mais simples, aquela que necessita menor número de alterações de caráteres.Cada ramo de uma árvore filogenética deve ser sustentado por pelo menos uma sinapomorfia, um estado derivado do traço compartilhado por todas as espécies.Essa técnica descreve quais estados de caracteres são primitivos e quais são derivados.Não existe nenhum grupo primitivo ou derivado.Um sapo não é "primitivo" em relação a répteis e mamíferos, ele é uma espécie totalmente moderna, mas que reteve alguns traços primitivos dos ancestrais de répteis e mamíferos que muito provavelmente existiram no Devoniano.
Assim, as sinapomorfias são vistas em estudos comparativos bioquímicos, genéticos, embriológicos e morfológicos.Traços comuns podem ser vistos através de homologias e de estruturas vestigiais.Homologia significa uma mesma estrutura em diversos organismos que tem uma variedade de formas e funções.Para a sistemática filogenética, ela significa também uma hipótese que pode ser testada e existe apenas quando a estrutura deriva de um ancestral comum(ver parte 6 deste artigo). Quando a estrutura existe devido a evolução convergente, reversão evolutiva ou evolução paralela é chamada de homoplasia.Por fim, estrutura vestigial é um caráter reduzido e incipientemente desenvolvido comparado a uma mesma estrutura complexa existente em outro organismo.
A evidência para a ancestralidade comum não começa com a construção de filogenias e a diferenciação de homologia ou homoplasia e sim com os testes de agrupamento de muitos traços. Eles mostram que os organismos agrupam naturalmente em uma hierarquia aninhada. Cada um dos táxons tem várias sinapomorfias que as distinguem de outros táxons. Traços não são facilmente misturados e igualados em grupos de organismos. Por exemplo, flores são vistas apenas em plantas que carregam vários outros caracteres que as distinguem como angiospermas.Todas as espécies em um grupo irão compartilhar traços herdados de seu ancestral comum. Mas, cada grupo e subgrupo (de espécies até táxons superiores como filos) terá evoluído seus próprios traços únicos.Ocasionalmente, as gimnospermas poderiam ser encontradas com flores, mas não são.Alguns pássaros poderiam ser encontrados com glândulas ou pêlos, mas não são.Ter penas seria muito útil como isolante térmico em mamíferos, mas essas e outras características não existem devido à restrição de descendência.A idéia da estrutura hierárquica na cladística pode ser comprovada estatisticamente através do índice de consistência (Klassen, 1991).O índice de consistência é o número mínimo de passos que o caracter pode mostrar (m), dividido pelo número de passos que ele efetivamente mostra em um dado cladograma (s).Klassen testou o IC de 75 cladogramas.O valor do IC depende do número de táxons no cladograma.Para um CI ao redor de 0,2 para 20 táxons, é considerado uma correlação boa em termos estatísticos.Mas se ele estivesse abaixo de 0,1 para esse número de táxons , seria o indicativo de uma estrutura anti-cladística (Theobald,2004). Encontrar um organismo que misturasse facilmente traços de mais de um táxon, como um animal metade-mamífero, metade-ave destruiria a hipótese da estrutura hierárquica cladística que é confirmada estatisticamente.Entretanto, como um animal desse tipo nunca é encontrado, e esse padrão sugere descendência comum.Existem agrupamentos de similaridades que não sugerem isso.As similaridades utilizadas em fabricações de automóveis seguem o padrão do gosto de um consumidor, mas não se agrupam hierarquicamente, porque os traços são facilmente igualados nos grupos e nos subgrupos.Agora, os vários idiomas do mundo estarão aproximadamente agrupados hierarquicamente, porque existe uma semelhança tênue com o aninhamento que ocorre na vida.Agrupar organismos hierarquicamente foi praticado primeiramente por Linnaeus, um criacionista.Isso mostra que esse tipo de agrupamento não é uma suposição evolucionista.Agrupar sugerindo muitos traços comuns é a principal evidência da ancestralidade comum.
Além disso, é necessário esclarecer o que é “representar a história da vida no gráfico da árvore” :construir uma filogenia e a ordem da sequência evolutiva da filogenia . A filogenia é uma hipótese acerca das relações como uma árvore genealógica, independente de a árvore está "enraizada" ou não.E a filogenia "enraizada", é a genealogia com a seqüência evolutiva especificada na árvore, ou seja, é uma filogenia com uma árvore "enraizada", geralmente tendo um grupo externo sobre controle.O grupo externo geralmente é um grupo irmão dos táxons comparados.Por exemplo, um peixe seria um grupo externo de uma filogenia de tetrápodes.Numa árvore filogenética, todas as partes são inferidas, exceto pelas dos ramos, as quais são espécies observadas e descritas, viventes ou fósseis.Espécies fósseis são usualmente interpretados como ponta de ramos extintos, não como ancestrais viventes das espécies atuais. Filogenias da cladística são baseadas na morfologia.Assim, se tem uma filogenia bem-suportada, a ordem em que os organismos são requeridos para surgir são preditos detalhadamente baseados nessa filogenia, e dados distintos (moleculares, paleontológicos, etc.) devem apresentar congruência com ela, caso ela seja verdadeira.
A maneira como se determina a cronologia dos ancestrais comuns está de acordo com o princípio da parcimônia.Podemos mostrar como funciona o critério da parcimônia na construção de árvores.A hipótese é que a evolução só faz sentido se proceder parcimoniosamente.Um exemplo, se se põe num cladograma, um urso, um chimpanzé e um homem e temos duas hipóteses: uma que agrupa o chimpanzé com o homem.E outra que agrupa o chimpanzé com o urso. As características semelhantes dos primatas são inovações. Inovações são eventos improváveis.Na primeira hipótese, as características semelhantes dos primatas se originam somente uma vez, e na segunda hipótese surgiram duas vezes.Utiliza-se o critério da parcimônia.A primeira árvore necessita menos inovações (mutações genéticas).E ela não é automaticamente verdadeira, mas é a melhor hipótese.Uma mudança evolucionária tal como as que envolvem novas características pode ser considerada como custo. Portanto, este método tenta encontrar o custo evolucionário mínimo de uma árvore se baseando nos estados derivados compartilhados por pares de táxons (é feita uma matriz dos estados de caracteres compartilhados).Para a evolução,a ameba é uma espécie tão moderna quanto nós.O que acontece é que o ancestral inferido pela evolução é unicelular, porque seria contra o princípio da parcimônia se a multicelularidade tivesse surgido várias vezes independentemente.Filogenias também refletem o grau de parentesco.Por exemplo, se mamíferos estão mais próximos entre si que peixes em grau de parentesco por linhas de evidência independentes, isso se deve ao fato que eles estão conectados pelo mesmo ancestral comum.
O método de construção de árvore da máxima parcimônia se aplica a dados morfológicos ou moleculares, ou ambos. Para seqüências de DNA com homoplasias usuais, geralmente são aplicados outros métodos, como métodos de distância e a máxima semelhança.No primeiro caso, a método começa pelo cálculo de distância entre todos os pares de espécies.No segundo, a árvore é encontrada considerando um grande conjunto de árvores candidatas hipotéticas e utilizando-se a estatística de um método computacional.
Existem diferentes métodos para calcular a confiabilidade de uma árvore.Um notável é o bootstrapping que faz isso por meio de softwares e da amostragem dos dados originais, calculando uma nova árvore com o banco de dados e repetindo o processo muitas vezes.Os valores de bootstrap são as porcentagens de casos nos quais os grupos aparecem na árvore resultante.Valores de bootstrap indicam que uma filogenia tem suporte quando um ramo é ótimo em 90% das subamostras e aqueles com valor abaixo de 70% são vistos com suspeitas (Stearns & Hoekstra,2003.p.249).Isso mostra que algumas filogenias são menos confiáveis porque os dados são igualmente consistentes com diversos padrões de ramificação.Só as que tem forte suporte representam uma descrição realista e rigorosa da descendência comum.Tais técnicas cladísticas não são coisas abstratas que nunca detectam árvores verdadeiras e sem utilidade prática.Elas já demonstraram que um dentista transmitiu HIV para seus pacientes, comparando os genomas dos vírus das pessoas infectadas e de suspeitos (Hillis et al,1996).Há outro caso onde a filogenia com 135.135 árvores possíveis de bacteriófagos que se propagaram na presença de mutágenos, a árvore verdadeira foi apontada indepentemente por cinco diferentes métodos (Hillis et al., 1992).
Como são calculadas as discrepâncias das filogenias? Tem um sub-artigo do já citado artigo de Douglas Theobald "29+ Evidences for Macroevolution: The Scientific Case for Common Descent" intitulado "Some Statistics of Incongruent Phylogenetic Trees" que mostra como é calculada a congruência estatística desses dois dados independentes.O valor de P, mede a incongruência estatística entre árvores independentes.Ela é a razão do número máximo de árvores inconsistentes sobre o número total de árvores possíveis.Quem quiser calcular, é só ir a esse link na seção "P-value Calculator", digitar o número dois em "Number of Trees" (número de árvores comparadas), digitar o número de táxons envolvidos na árvore em "Number of Taxa" e o número de ramos de cada árvore que se tem que "cortar" para que as duas fiquem idênticas em "Number of Incongruent Branches".
No artigo de Theobald é mostrado a análise das filogenias dos crocodilianos (envolvendo oito táxons) construídas com dados morfológicos e moleculares.Ela tem um valor de P de 0,000769. Dado que existem 135.135 árvores possíveis, seria muito fácil que as duas árvores não convergissem , mas só há um único ramo inconsistente.
Segundo o artigo, existe elevada significância estatística quando P é menor que 0,01; significância estatística, quando P é menor que 0,05; valor equívoco, quando P está entre 0,05 e 0,50; e valor altamente insignificante, quando P é maior que 0,50.
Referências:
Hillis, D.M., J.J. Bull, M.E. White, M.R. Badgett and I. J. Molineux (1992).Experimental phylogenetics: Generation of a known phylogeny.Science 255: 589-592.
Hillis,D.M.,Moritz,C. and Mable, B.K.(1996).Molecular systematics, (2th edn). Sinauer Associates, Sunderland, Massaschsetts.
Klassen, G. J., Mooi, R. D., and Locke, A. (1991) Consistency indices and random data. Syst. Zool. 40:446-457.
Mayr,E. (2005).Biologia, Ciência Única.Editora Companhia das Letras, São Paulo-SP.
Pazza, R. (2004) Espécies em anel. Projeto Evoluindo - Biociência.org. [http://www.evoluindo.biociencia.org/ringspecies.htm]
Ridley, M. (2006).Evolução, 3 ª edição, Editora Artmed, Porto Alegre-RS.
Standfast, A. L. (1992).The human tail.New York State Journal of Medicine. 92: 116.
Stearns S.C. e Hoekstra, R.F. (2003).Evolução: Uma Introdução.Atheneu Editora São Paulo, São Paulo-SP.
Theobald, Douglas L. “29+ Evidences for Macroevolution: The Scientific Case for Common Descent." The Talk.Origins Archive. Vers. 2.83. 2004. 12 Jan, 2004 <http://www.talkorigins.org/faqs/comdesc/>
Weinberg, J.R., Starczak,V.R. and Jorg, D.(1992).Evidence for rapid speciation following a founder event in the laboratory. Evolution 46: 1214-1220.
---------------------------------------------------------------------------------------------------------------------------------------
A descendência com modificação não foi apenas observada apenas dentro da espécie, mas também em relações interespecíficas.A macroevolução já foi observada diretamente e indiretamente.Origem de uma nova espécie pela evolução já foi vista até em laboratório (Weinberg, et al., 1992) e um tipo de especiação merece atenção porque permite um estudo detalhado. É o caso do fenômeno “espécies-em-anel”, onde vemos diferenças devido a isolamento parcial dando origem a populações que se comportam como espécies separadas.É uma situação na qual duas populações que vivem em um mesma região e não se reproduzem estão conectadas por um anel geográfico de populações que se reproduzem.Este é o caso do pássaro Phylloscopus trochiloides e da salamandra do gênero Ensatina (Pazza,2004).A última possui tantas diferenças, que duas populações desenvolveram estratégias radicalmente diferentes para evitar predadores-uma exibe padrão críptico e outra mimetiza uma espécie de salamandra venenosa.Além disso, existem as evidências da descendência comum baseada em observação indireta. Elas incluem: anatomia comparativa, biologia do desenvolvimento comparativa, estudos comparativos bioquímicos e genéticos; padrões de biogeografia e o registro fóssil.
O que está por trás da afirmação "que todos os seres descendam gradualmente modificados de uma forma ou de algumas formas originais"?O gradualismo não é um mecanismo de mudança evolutiva.E também não é uma afirmação quanto a taxas ou tempo da evolução (Mayr,2005.p.122).Eventos geneticamente graduais são apenas restrições da área de variação biológica esperada entre duas gerações quaisquer quando observamos suas taxas genéticas, taxas morfológicas,etc. Assim, o gradualismo requer apenas restrição a possíveis mecanismos de descendência comum e adaptação, não afirmando nada sobre se a evolução deve ser constante,rápida, ocorrer frequentemente,etc.Acreditar que é "discutível que os seres descendem de um ancestral comum", porque não ser possível a história da vida esteja "representada pelo gráfico da árvore" mostra apenas uma confusão no entendimento de conceitos científicos.É análogo a dizer que "é discutível que a força entre duas massas sempre esteja de acordo com a lei do quadrado do inverso.Se assim fosse, a aceleração de todas as massas estariam de acordo com a existência de um valor exato da constante gravitacional G."A razão de seu comentário está incorreto é que a construção de uma "representação da vida no gráfico da árvore" não é possível, porque valores exatos de mensurações não são sabidos em ciência.Aproximações de mensurações não existem apenas em biologia evolutiva, mas em todas as ciências.O que não é impossível é construir uma árvore filogenética padrão, uma aproximação da verdadeira e única genealogia da vida.O mesmo poderíamos dizer sobre o conhecimento do valor exato da constante gravitacional universal, já que até este tem medidas independentes que desacordam.O que se pode fazer é construir uma constante gravitacional G padrão para que todas as massas sempre se acelerem em um valor específico que nunca seja massivamente inconsistente com ela.De modo similar, o que se pode fazer para testar a descendência comum universal é construir uma árvore filogenética padrão, onde toda filogenia específica seja compatível com ela.
Árvores são construídas a partir de dados, com a utilização de métodos que podem produzir árvores distintas.Se os dados são igualmente consistentes com diversos padrões de ramificação, o julgamento acerca das relações deve ser suspenso.Mas uma filogenia bem determinada pode realizar as predições da teoria da descendência comum universal se todos os métodos devem fornecer o mesmo ramo em um grande (e confiável) conjunto de dados e com cada ramo sendo bem sustentado.Por fim, considerando que qualquer filogenia com tais características é a melhor aproximação de uma verdadeira e única genealogia de todas as espécies, ela forma uma árvore filogenética padrão.Isso significa que apenas uma filogenia assim pode ser uma descrição realista e rigorosa da descendência comum.Um exemplo prático dela é esta árvore neste gráfico criado pelo bioquímico norte-americano Douglas Theobald (2004) no Talk Origins Archive.
Outra questão no teste de filogenias é explicar porque ela não prediz a exata congruência de mensurações filogenéticas construídas por dados independentes (seja por dados moleculares, embriológicos, paleontológicos e/ou morfológicos).Em caso de filogenias envolvendo muitos táxons, o simples fato de colocarmos muitas espécies num cladograma faz com que o número de árvores filogenéticas possíveis cresça fatorialmente, porque a fórmula para se calcular o número de árvores possíveis é NR=(2n-3)!! ou (2n-3)!/(2n-2(n-2)!), onde "n" é o número de táxons no cladograma.No caso de cinco espécies são 105 árvores possíveis.Em caso de 10, são 34.459.425.No caso de 15 espécies, são 213.458.046.676.875 árvores possíveis.Mas se se consegue reduzir o número de árvores prováveis para 34 , isso significa que se está fazendo uma mensuração com uma acurácia de uma parte em 1 milhão (34/34.459.425 árvores possíveis) .Se colocássemos 15 espécies,poderia-se estabelecer centenas de árvores prováveis e a acurácia seria considerada ainda maior.Como não existe mensuração perfeita na obtenção de valores independentes em ciência, cientistas utilizam estatísticas de análise de congruência , para estimar o quanto de discrepância a teoria permite para conflitos de árvores filogenéticas obtidas por dados independentes (sejam eles moleculares, embriológicos, morfológicos, etc.).Um dos cálculos de incongruência filogenética é o valor de P e o método mais popular de avaliação da confiabilidade de árvores é o bootstrapping (ambos serão analisados a seguir).Como se vê na parte 5 deste artigo, a evidência disponível existe um número excepcional de filogenias bem determinadas inferidas por métodos independentes combinando-se com significância estatística.Outra coisa deve ser analisada.Se a descendência comum fosse falsa, existiriam independentes filogenias com forte suporte mostrando nível de incongruência elevada.Como isso ainda não ocorre, percebe-se que ainda não surgiu dados que parecessem um problema sério para a teoria.
Se a descendência com modificação é aceitável dentro da espécie, porque não em relações intraespecíficas?Nunca observamos diretamente que todos os humanos descendendo gradualmente de um ancestral remoto, mas temos evidências impressionantes disso.Idem para a descendência com modificação em relações inter-específicas.
Algumas filogenias são consensuais e tem ramos bem-suportados, de modo que uma inversão nela refutaria a evolução.Por exemplo, a evolução prediz com certeza que humanos já tiveram ancestrais caudados,assim como os primeiros primatas.Taxonomistas englobam humanos, chimpanzés, gorilas, orangotangos e gibões na Superfamília Homonoidea e são justamente esses animais que compartilham a sinapomorfia "ausência de cauda".Podemos citar um exemplo de uma evidência que confirma uma hipótese da filogenia de consenso universal e outro que a contradiga?Sim, a existência de atavismos em bebês humanos que possuem caudas , fato registrado na literatura médica (ver fotos no link).Como isso pode ocorrer? Na cauda embrionária, existente em nós humanos, e em outros macacos, em determinado estágio essas células morrem.Porém, uma mutação faz com que as células da cauda morram um tempo após terem sido formadas. Da mesma forma que uma mutação fixada na maioria da população é responsável pela ausência da cauda na maioria de nós, mutações raras podem fazê-las reaparecer em alguns indivíduos.Os primeiros primatas tinham caudas e a ausência delas é uma característica derivada que é existente em apenas em cinco grupos primatas que representam a Superfamília Hominoidea.Vértebras e cartilagem têm ocasionalmente sido encontradas em caudas humanas. De qualquer forma, vértebras não são um requerimento para caudas. M. sylvanus é um exemplo perfeito de primata cuja cauda carnuda não têm vértebras. Nós agora sambemos os genes responsáveis pelo desenvolvimento das caudas em mamíferos, e que todos os humanos os têm. Muita dessas caudas são complexas estruturas compostas de camadas simétricas de músculos voluntários, vasos sangüíneos, nervos especializados e órgãos sensitivos, são raramente herdadas, ainda que vários casos familiares sejam conhecidos. Em um caso isso foi herdado por pelo menos três gerações de indivíduos do sexo feminino. (Standfast,1992).Não seria indício de ancestralidade comum se fossem homoplasias, mas não são. Os genes existem e produzem a cauda que permanece em todos os animais até um certo período de gestação, quando nos primatas mais modernos elas regridem.Baseado na hipótese da descendência comum e na árvore filogenética padrão , podemos esperar ver caudas atávicas em humanos, mas essa hipótese poderia ser confirmada como falsa se víssemos algum caso de bebê nascer com escamas.
Há evidência até para o caso em que todos estão agrupados num único grande táxon universal.Por exemplo, sabe-se que todos os seres dos cinco reinos tem um genoma baseado em ácidos nucléicos e o mesmo código genético que usam os mesmos códons e aminoácidos porque descendem de um ancestral comum, poderia se encontrar uma evidência contrária falseadora se descobrissem algum organismo com um genoma que não é baseado em ácidos nucléicos ou que utilize o mesmo código genético com códons e aminoácidos diferentes.Uma vez que ter exatamente as mesmas trincas que codificam exatamente os mesmos aminoácidos não é funcionalmente necessário e nem se deve a razões de afinidades químicas , a explicação mais racional é que ele é um "acidente congelado",onde um desvio seria letal.Um organismo que lesse GGC como fenilalanina em vez de glicina, morreria ainda no estágio de ovo (Ridley,2006.p.81).
Por fim, é bom esclarecer que a teoria da descendência comum universal é a única que pode dar conta de explicar cientificamente da unidade, diversidade, e padrões da vida terrestre.Ela pode ser testada independentemente dos mecanismos teóricos.Ela também é considerada fato científico pela maioria dos biólogos, embora algumas teorias evolutivas sobre os mecanismos evolutivos possam não ter o mesmo status.
COMO AS FILOGENIAS SÃO CONSTRUÍDAS E AVALIADAS?
Para entender como a descendência comum é descrita pelas filogenias é necessário compreender a metodologia da sistemática filogenética.A biologia evolutiva está constantemente tentando encontrar grupos monofiléticos.Quando um grupo é considerado monofilético, isso quer dizer que todos os ancestrais daquele grupo são derivados de um ancestral comum, o qual não é ancestral de nenhum outro grupo.A abordagem a sistemática, conhecida atualmente como cladística, foi formulada por Willi Hennig e estabelece que as sinapomorfias - estados de caráter unicamente compartilhados e derivados- são filogeneticamente informativas.Assim, se compararmos o traço “número de dígitos”, ele não seria relevante numa análise filogenética entre humanos, lagartos e cavalos, já que é ancestral a todos os três.Mas se considerarmos cavalos, zebras e humanos ele seria relevante, pois se encontra num estado derivado por cavalos e zebras em relação a humanos.A similaridade primitiva herdada de um ancestral distante é chamada de plesiomorfia.As que são derivadas são chamadas de apomorfias.Uma sinapomorfia em níveis taxonômicos mais altos pode representar uma plesiomorfia desinteressante em níveis inferiores.Um exoesqueleto quintinoso ajuda a distinguir artrópodes de anelídeos e dos moluscos, mas não é útil na distinção entre espécies de borboletas.Se as espécies A, B e C reunem mais traços comuns entre si que D, E e F (e com um postulado ancestral comum mais recente por causa disso), então pelo critério da parcimônia, uma característica em comum compartilhada por D,E e F é provavelmente ancestral.É possível a codificação dos caracteres, de forma que os mesmos possam ser utilizados em um algoritmo, manual e automatizado.Procura-se a árvore mais simples, aquela que necessita menor número de alterações de caráteres.Cada ramo de uma árvore filogenética deve ser sustentado por pelo menos uma sinapomorfia, um estado derivado do traço compartilhado por todas as espécies.Essa técnica descreve quais estados de caracteres são primitivos e quais são derivados.Não existe nenhum grupo primitivo ou derivado.Um sapo não é "primitivo" em relação a répteis e mamíferos, ele é uma espécie totalmente moderna, mas que reteve alguns traços primitivos dos ancestrais de répteis e mamíferos que muito provavelmente existiram no Devoniano.
Assim, as sinapomorfias são vistas em estudos comparativos bioquímicos, genéticos, embriológicos e morfológicos.Traços comuns podem ser vistos através de homologias e de estruturas vestigiais.Homologia significa uma mesma estrutura em diversos organismos que tem uma variedade de formas e funções.Para a sistemática filogenética, ela significa também uma hipótese que pode ser testada e existe apenas quando a estrutura deriva de um ancestral comum(ver parte 6 deste artigo). Quando a estrutura existe devido a evolução convergente, reversão evolutiva ou evolução paralela é chamada de homoplasia.Por fim, estrutura vestigial é um caráter reduzido e incipientemente desenvolvido comparado a uma mesma estrutura complexa existente em outro organismo.
A evidência para a ancestralidade comum não começa com a construção de filogenias e a diferenciação de homologia ou homoplasia e sim com os testes de agrupamento de muitos traços. Eles mostram que os organismos agrupam naturalmente em uma hierarquia aninhada. Cada um dos táxons tem várias sinapomorfias que as distinguem de outros táxons. Traços não são facilmente misturados e igualados em grupos de organismos. Por exemplo, flores são vistas apenas em plantas que carregam vários outros caracteres que as distinguem como angiospermas.Todas as espécies em um grupo irão compartilhar traços herdados de seu ancestral comum. Mas, cada grupo e subgrupo (de espécies até táxons superiores como filos) terá evoluído seus próprios traços únicos.Ocasionalmente, as gimnospermas poderiam ser encontradas com flores, mas não são.Alguns pássaros poderiam ser encontrados com glândulas ou pêlos, mas não são.Ter penas seria muito útil como isolante térmico em mamíferos, mas essas e outras características não existem devido à restrição de descendência.A idéia da estrutura hierárquica na cladística pode ser comprovada estatisticamente através do índice de consistência (Klassen, 1991).O índice de consistência é o número mínimo de passos que o caracter pode mostrar (m), dividido pelo número de passos que ele efetivamente mostra em um dado cladograma (s).Klassen testou o IC de 75 cladogramas.O valor do IC depende do número de táxons no cladograma.Para um CI ao redor de 0,2 para 20 táxons, é considerado uma correlação boa em termos estatísticos.Mas se ele estivesse abaixo de 0,1 para esse número de táxons , seria o indicativo de uma estrutura anti-cladística (Theobald,2004). Encontrar um organismo que misturasse facilmente traços de mais de um táxon, como um animal metade-mamífero, metade-ave destruiria a hipótese da estrutura hierárquica cladística que é confirmada estatisticamente.Entretanto, como um animal desse tipo nunca é encontrado, e esse padrão sugere descendência comum.Existem agrupamentos de similaridades que não sugerem isso.As similaridades utilizadas em fabricações de automóveis seguem o padrão do gosto de um consumidor, mas não se agrupam hierarquicamente, porque os traços são facilmente igualados nos grupos e nos subgrupos.Agora, os vários idiomas do mundo estarão aproximadamente agrupados hierarquicamente, porque existe uma semelhança tênue com o aninhamento que ocorre na vida.Agrupar organismos hierarquicamente foi praticado primeiramente por Linnaeus, um criacionista.Isso mostra que esse tipo de agrupamento não é uma suposição evolucionista.Agrupar sugerindo muitos traços comuns é a principal evidência da ancestralidade comum.
Além disso, é necessário esclarecer o que é “representar a história da vida no gráfico da árvore” :construir uma filogenia e a ordem da sequência evolutiva da filogenia . A filogenia é uma hipótese acerca das relações como uma árvore genealógica, independente de a árvore está "enraizada" ou não.E a filogenia "enraizada", é a genealogia com a seqüência evolutiva especificada na árvore, ou seja, é uma filogenia com uma árvore "enraizada", geralmente tendo um grupo externo sobre controle.O grupo externo geralmente é um grupo irmão dos táxons comparados.Por exemplo, um peixe seria um grupo externo de uma filogenia de tetrápodes.Numa árvore filogenética, todas as partes são inferidas, exceto pelas dos ramos, as quais são espécies observadas e descritas, viventes ou fósseis.Espécies fósseis são usualmente interpretados como ponta de ramos extintos, não como ancestrais viventes das espécies atuais. Filogenias da cladística são baseadas na morfologia.Assim, se tem uma filogenia bem-suportada, a ordem em que os organismos são requeridos para surgir são preditos detalhadamente baseados nessa filogenia, e dados distintos (moleculares, paleontológicos, etc.) devem apresentar congruência com ela, caso ela seja verdadeira.
A maneira como se determina a cronologia dos ancestrais comuns está de acordo com o princípio da parcimônia.Podemos mostrar como funciona o critério da parcimônia na construção de árvores.A hipótese é que a evolução só faz sentido se proceder parcimoniosamente.Um exemplo, se se põe num cladograma, um urso, um chimpanzé e um homem e temos duas hipóteses: uma que agrupa o chimpanzé com o homem.E outra que agrupa o chimpanzé com o urso. As características semelhantes dos primatas são inovações. Inovações são eventos improváveis.Na primeira hipótese, as características semelhantes dos primatas se originam somente uma vez, e na segunda hipótese surgiram duas vezes.Utiliza-se o critério da parcimônia.A primeira árvore necessita menos inovações (mutações genéticas).E ela não é automaticamente verdadeira, mas é a melhor hipótese.Uma mudança evolucionária tal como as que envolvem novas características pode ser considerada como custo. Portanto, este método tenta encontrar o custo evolucionário mínimo de uma árvore se baseando nos estados derivados compartilhados por pares de táxons (é feita uma matriz dos estados de caracteres compartilhados).Para a evolução,a ameba é uma espécie tão moderna quanto nós.O que acontece é que o ancestral inferido pela evolução é unicelular, porque seria contra o princípio da parcimônia se a multicelularidade tivesse surgido várias vezes independentemente.Filogenias também refletem o grau de parentesco.Por exemplo, se mamíferos estão mais próximos entre si que peixes em grau de parentesco por linhas de evidência independentes, isso se deve ao fato que eles estão conectados pelo mesmo ancestral comum.
O método de construção de árvore da máxima parcimônia se aplica a dados morfológicos ou moleculares, ou ambos. Para seqüências de DNA com homoplasias usuais, geralmente são aplicados outros métodos, como métodos de distância e a máxima semelhança.No primeiro caso, a método começa pelo cálculo de distância entre todos os pares de espécies.No segundo, a árvore é encontrada considerando um grande conjunto de árvores candidatas hipotéticas e utilizando-se a estatística de um método computacional.
Existem diferentes métodos para calcular a confiabilidade de uma árvore.Um notável é o bootstrapping que faz isso por meio de softwares e da amostragem dos dados originais, calculando uma nova árvore com o banco de dados e repetindo o processo muitas vezes.Os valores de bootstrap são as porcentagens de casos nos quais os grupos aparecem na árvore resultante.Valores de bootstrap indicam que uma filogenia tem suporte quando um ramo é ótimo em 90% das subamostras e aqueles com valor abaixo de 70% são vistos com suspeitas (Stearns & Hoekstra,2003.p.249).Isso mostra que algumas filogenias são menos confiáveis porque os dados são igualmente consistentes com diversos padrões de ramificação.Só as que tem forte suporte representam uma descrição realista e rigorosa da descendência comum.Tais técnicas cladísticas não são coisas abstratas que nunca detectam árvores verdadeiras e sem utilidade prática.Elas já demonstraram que um dentista transmitiu HIV para seus pacientes, comparando os genomas dos vírus das pessoas infectadas e de suspeitos (Hillis et al,1996).Há outro caso onde a filogenia com 135.135 árvores possíveis de bacteriófagos que se propagaram na presença de mutágenos, a árvore verdadeira foi apontada indepentemente por cinco diferentes métodos (Hillis et al., 1992).
Como são calculadas as discrepâncias das filogenias? Tem um sub-artigo do já citado artigo de Douglas Theobald "29+ Evidences for Macroevolution: The Scientific Case for Common Descent" intitulado "Some Statistics of Incongruent Phylogenetic Trees" que mostra como é calculada a congruência estatística desses dois dados independentes.O valor de P, mede a incongruência estatística entre árvores independentes.Ela é a razão do número máximo de árvores inconsistentes sobre o número total de árvores possíveis.Quem quiser calcular, é só ir a esse link na seção "P-value Calculator", digitar o número dois em "Number of Trees" (número de árvores comparadas), digitar o número de táxons envolvidos na árvore em "Number of Taxa" e o número de ramos de cada árvore que se tem que "cortar" para que as duas fiquem idênticas em "Number of Incongruent Branches".
No artigo de Theobald é mostrado a análise das filogenias dos crocodilianos (envolvendo oito táxons) construídas com dados morfológicos e moleculares.Ela tem um valor de P de 0,000769. Dado que existem 135.135 árvores possíveis, seria muito fácil que as duas árvores não convergissem , mas só há um único ramo inconsistente.
Segundo o artigo, existe elevada significância estatística quando P é menor que 0,01; significância estatística, quando P é menor que 0,05; valor equívoco, quando P está entre 0,05 e 0,50; e valor altamente insignificante, quando P é maior que 0,50.
Referências:
Hillis, D.M., J.J. Bull, M.E. White, M.R. Badgett and I. J. Molineux (1992).Experimental phylogenetics: Generation of a known phylogeny.Science 255: 589-592.
Hillis,D.M.,Moritz,C. and Mable, B.K.(1996).Molecular systematics, (2th edn). Sinauer Associates, Sunderland, Massaschsetts.
Klassen, G. J., Mooi, R. D., and Locke, A. (1991) Consistency indices and random data. Syst. Zool. 40:446-457.
Mayr,E. (2005).Biologia, Ciência Única.Editora Companhia das Letras, São Paulo-SP.
Pazza, R. (2004) Espécies em anel. Projeto Evoluindo - Biociência.org. [http://www.evoluindo.biociencia.org/ringspecies.htm]
Ridley, M. (2006).Evolução, 3 ª edição, Editora Artmed, Porto Alegre-RS.
Standfast, A. L. (1992).The human tail.New York State Journal of Medicine. 92: 116.
Stearns S.C. e Hoekstra, R.F. (2003).Evolução: Uma Introdução.Atheneu Editora São Paulo, São Paulo-SP.
Theobald, Douglas L. “29+ Evidences for Macroevolution: The Scientific Case for Common Descent." The Talk.Origins Archive. Vers. 2.83. 2004. 12 Jan, 2004 <http://www.talkorigins.org/faqs/comdesc/>
Weinberg, J.R., Starczak,V.R. and Jorg, D.(1992).Evidence for rapid speciation following a founder event in the laboratory. Evolution 46: 1214-1220.
posted by espectro109 at 5:13 PM
![]()

{kind=link}
1 Comments:
geotorelxz credit consolidation
low rate loans
debt consolidation loans
Post a Comment
<< Home